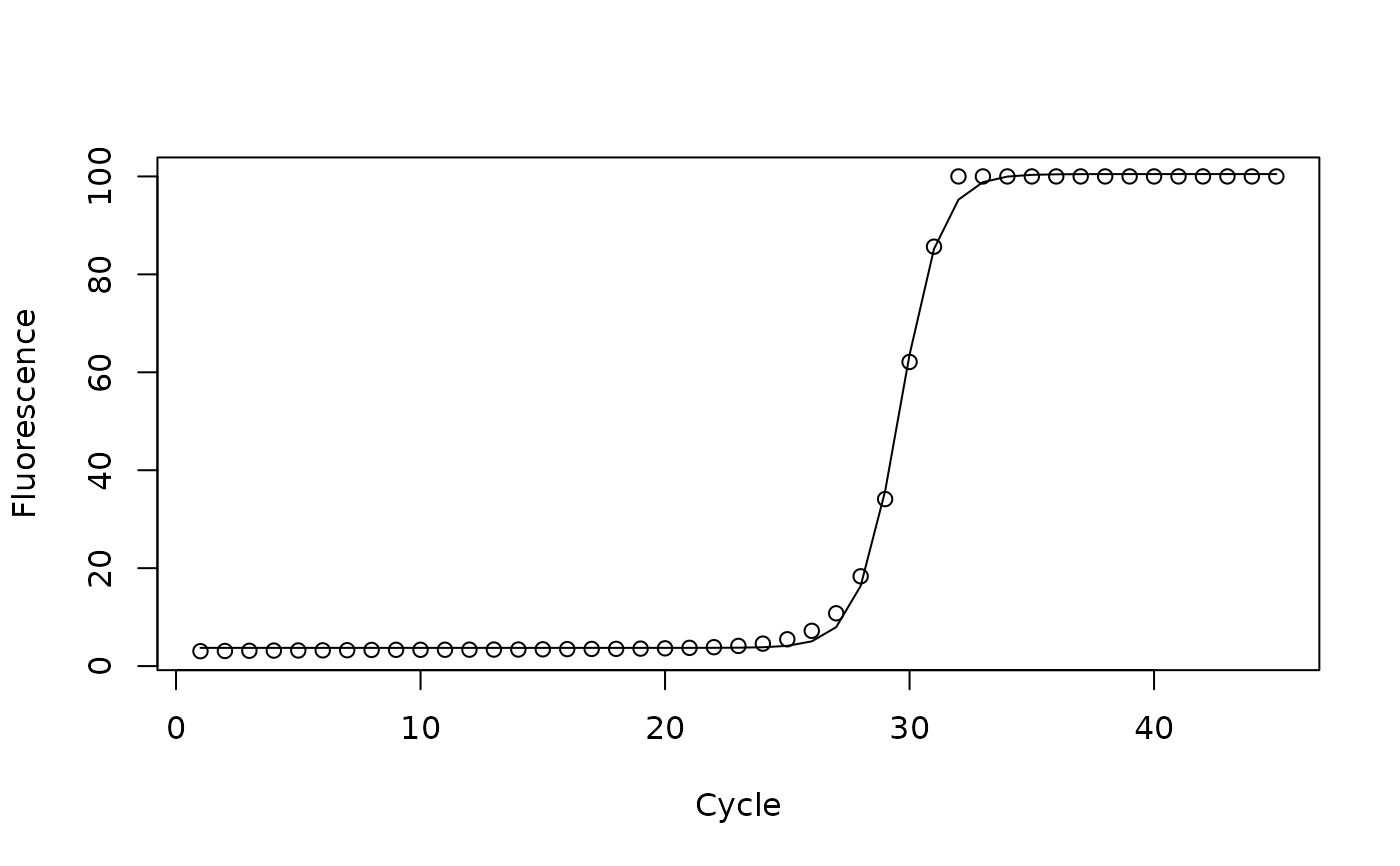
Gene expression levels from real-time quantitative polymerase chain reaction (qPCR) experiments on two different plant lines. Each line was used for 7 experiments each with 45 cycles.
Format
A data frame with 630 observations on the following 4 variables.
flour | numeric | Fluorescence level |
line | factor | Plant lines rnt (mutant) and wt
(wildtype) |
cycle | numeric | Cycle number for the experiment |
transcript | factor | Transcript used for the different runs |
Source
Data provided by Kirsten Jorgensen <kij@life.ku.dk>.
Added by Claus Ekstrom <ekstrom@life.ku.dk>
References
Morant, M. et al. (2010). Metabolomic, Transcriptional, Hormonal and Signaling Cross-Talk in Superroot2. Molecular Plant. 3, p.192–211.
Examples
data(qpcr)
#
# Analyze a single run for the wt line, transcript 1
#
run1 <- subset(qpcr, transcript==1 & line=="wt")
model <- nls(flour ~ fmax/(1+exp(-(cycle-c)/b))+fb,
start=list(c=25, b=1, fmax=100, fb=0), data=run1)
print(model)
#> Nonlinear regression model
#> model: flour ~ fmax/(1 + exp(-(cycle - c)/b)) + fb
#> data: run1
#> c b fmax fb
#> 29.5932 0.8406 96.7559 3.7226
#> residual sum-of-squares: 53.79
#>
#> Number of iterations to convergence: 9
#> Achieved convergence tolerance: 7.737e-06
plot(run1$cycle, run1$flour, xlab="Cycle", ylab="Fluorescence")
lines(run1$cycle, predict(model))